

{kind=link}
(The under publish accommodates excerpts from Slithering your method into
bioinformatics with snakemake, Hackmd Press, 2023.)
Set up and setup!
Setup and set up
I counsel working in a brand new listing.
You will must set up snakemake and sourmash. We recommend utilizing mamba, through miniforge/mambaforge, for this.
Getting the information:
You will must obtain these three information:
and rename them in order that they’re in a subdirectory genomes/ with the names:
GCF_000017325.1.fna.gz
GCF_000020225.1.fna.gz
GCF_000021665.1.fna.gz
Notice, you possibly can obtain saved copies of them right here, with the proper names: osf.io/2g4dm/.
Chapter 1 – snakemake runs applications for you!
Bioinformatics typically entails working many alternative applications to characterize and scale back sequencing information, and I exploit snakemake to assist me try this.
A primary, easy snakemake workflow
This is a easy, helpful snakemake workflow:
rule compare_genomes:
message: "evaluate all enter genomes utilizing sourmash"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
sourmash plot evaluate.mat
"""
Put it in a file referred to as Snakefile, and run it with snakemake -j 1.
It will produce the output file evaluate.mat.matrix.png which accommodates a similarity matrix and a dendrogram of the three genomes (see Determine 1).
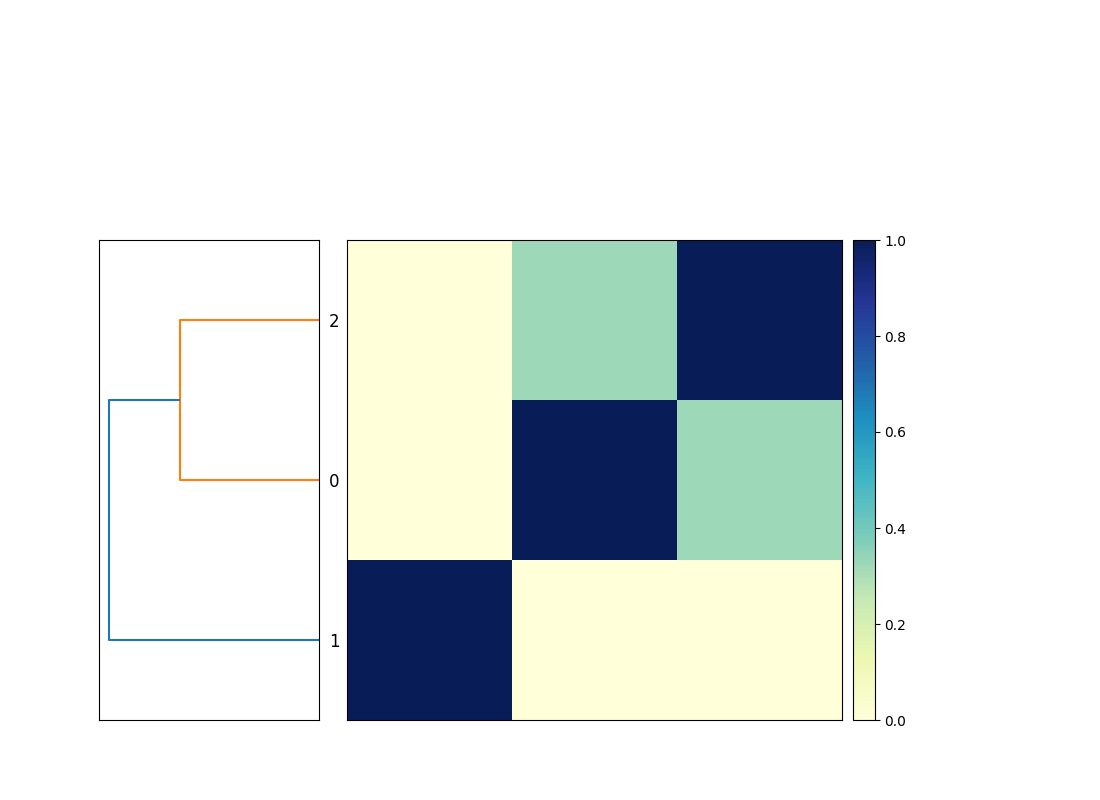
That is functionally equal to placing these three instructions right into a file compare-genomes.sh and working it with bash compare-genomes.sh –
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
sourmash plot evaluate.mat
The snakemake model is already somewhat bit nicer as a result of it would
provide you with encouragement when the instructions run efficiently (with good
inexperienced textual content saying “1 of 1 steps (100%) accomplished”!) and if the instructions
fail you will get purple textual content alerting you to that, too.
However! We will additional enhance the snakemake model over the shell
script model!
Avoiding pointless rerunning of instructions: a second snakemake workflow
The instructions will run each time you invoke snakemake with snakemake -j 1. However more often than not you needn’t rerun them since you’ve already received the output information you needed!
How do you get snakemake to keep away from rerunning guidelines?
We will try this by telling snakemake what we count on the output to be by including an output: block in entrance of the shell block:
rule compare_genomes:
message: "evaluate all enter genomes utilizing sourmash"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
sourmash plot evaluate.mat
"""
and now once we run snakemake -j 1 as soon as, it would run the instructions; however once we run it once more, it would say, “Nothing to be accomplished (all requested information are current and updated).”
It is because the specified output file, evaluate.mat.matrix.png, already exists. So snakemake is aware of it would not must do something!
In the event you take away evaluate.mat.matrix.png and run snakemake -j 1 once more, snakemake will fortunately make the information once more:
rm evaluate.mat.matrix.png
snakemake -j 1
So snakemake makes it simple to keep away from re-running a set of instructions if it
has already produced the information you needed. This is among the finest
causes to make use of a workflow system like snakemake for working
bioinformatics workflows; shell scripts do not routinely keep away from
re-running instructions.
Working solely the instructions it’s essential to run
The final Snakefile above has three instructions in it, however if you happen to take away the evaluate.mat.matrix.png file you solely must run the final command once more – the information created by the primary two instructions exist already and do not should be recreated. Nonetheless, snakemake would not know that – it treats the whole rule as one rule, and would not look into the shell command to work out what it would not must run.
If we wish to keep away from re-creating the information that exist already, we have to make the Snakefile somewhat bit extra sophisticated.
First, let’s escape the instructions into three separate guidelines.
rule sketch_genomes:
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
shell: """
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
"""
rule plot_comparison:
message: "evaluate all enter genomes utilizing sourmash"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash plot evaluate.mat
"""
We did not do something too sophisticated right here – we made two new rule blocks, with their very own names, and cut up the shell instructions up so that every shell command has its personal rule block.
You may inform snakemake to run all three:
snakemake -j 1 sketch_genomes compare_genomes plot_comparison
and it’ll efficiently run all of them!
Nonetheless, we’re again to snakemake working a number of the instructions each time – it will not run plot_comparison each time, as a result of evaluate.mat.matrix.png exists, however it would run sketch_genomes and compare_genomes repeatedly.
How will we repair this?
Including output blocks to every rule
If add output blocks to every rule, then snakemake will solely run guidelines
the place the output must be up to date (e.g. as a result of it would not exist).
Let’s try this:
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
output:
"evaluate.mat"
shell: """
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
"""
rule plot_comparison:
message: "evaluate all enter genomes utilizing sourmash"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash plot evaluate.mat
"""
and now
snakemake -j 1 sketch_genomes compare_genomes plot_comparison
will run every command solely as soon as, so long as the output information are nonetheless there. Huzzah!
However we nonetheless must specify the names of all three guidelines, in the proper order, to run this. That is annoying! Let’s repair that subsequent.
Chapter 2: snakemake connects guidelines for you!
Chaining guidelines with enter: blocks
We will get snakemake to routinely join guidelines by offering
details about the enter information a rule wants. Then, if you happen to ask
snakemake to run a rule that requires sure inputs, it would
routinely determine which guidelines produce these inputs as their
output, and routinely run them.
Let’s add enter data to the plot_comparison and compare_genomes
guidelines:
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
enter:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
output:
"evaluate.mat"
shell: """
sourmash evaluate GCF_000021665.1.fna.gz.sig
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig
-o evaluate.mat
"""
rule plot_comparison:
message: "evaluate all enter genomes utilizing sourmash"
enter:
"evaluate.mat"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash plot evaluate.mat
"""
Now you possibly can simply ask snakemake to run the final rule:
snakemake -j 1 plot_comparison
and snakemake will run the opposite guidelines provided that these enter information do not exist and should be created.
Taking a step again
The Snakefile is now loads longer, but it surely’s not too rather more sophisticated – what we have accomplished is cut up the shell instructions up into separate guidelines and annotated every rule with data about what file it produces (the output), and what information it requires with a view to run (the enter).
This has the benefit of constructing it so that you needn’t rerun instructions unnecessarily. That is solely a small benefit with our present workflow, as a result of sourmash is fairly quick. But when every step takes an hour to run, avoiding pointless steps could make your work go a lot quicker!
And, as you will see later, these guidelines are reusable constructing blocks that may be integrated into workflows that every produce totally different information. So there are different good causes to interrupt shell instructions out into particular person guidelines!
Chapter 3: snakemake helps you keep away from redundancy!
Avoiding repeated filenames through the use of {enter} and {output}
In the event you have a look at the earlier Snakefile, you will see a couple of repeated filenames – specifically, rule compare_genomes has three filenames within the enter block after which repeats them within the shell block, and evaluate.mat is repeated a number of instances in each compare_genomes and plot_genomes.
We will inform snakemake to reuse filenames through the use of {enter} and {output}. The { and } inform snakemake to interpret these not as literal strings however as template variables that must be changed with the worth of enter and output.
Let’s give it a strive!
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
enter:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
output:
"evaluate.mat"
shell: """
sourmash evaluate {enter} -o {output}
"""
rule plot_comparison:
message: "evaluate all enter genomes utilizing sourmash"
enter:
"evaluate.mat"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash plot {enter}
"""
This method not solely entails much less typing within the first place, but additionally makes it so that you simply solely must edit filenames in a single place. This avoids errors attributable to including or altering filenames in a single place and never one other place – a mistake I’ve made loads of instances!
snakemake makes it simple to rerun workflows!
It is not uncommon to wish to rerun a complete workflow from scratch, to just remember to’re utilizing the most recent information information and software program. Snakemake makes this simple!
You may ask snakemake to wash up all of the information that it is aware of the best way to generate – and solely these information:
snakemake -j 1 plot_comparison --delete-all-output
which might then be adopted by asking snakemake to regenerate the outcomes:
snakemake -j 1 plot_comparison
snakemake will provide you with a warning to lacking information if it could possibly’t make them!
Suppose you add a brand new file that doesn’t exist to compare_genomes:
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p okay=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
enter:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig",
"does-not-exist.sig"
output:
"evaluate.mat"
shell: """
sourmash evaluate {enter} GCF_000021665.1.sig -o {output}
"""
rule plot_comparison:
message: "evaluate all enter genomes utilizing sourmash"
enter:
"evaluate.mat"
output:
"evaluate.mat.matrix.png"
shell: """
sourmash plot {enter}
"""
Right here, does-not-exist.sig would not exist, and we’ve not given snakemake a rule to make it, both. What’s going to snakemake do??
It’s going to complain, loudly and clearly! And it’ll achieve this earlier than working something.
First, let’s drive the rule take away the output file that depends upon the
after which run snakemake -j 1. It is best to see:
Lacking enter information for rule compare_genomes:
output: evaluate.mat
affected information:
does-not-exist.sig
That is precisely what you need – a transparent indication of what’s lacking earlier than your workflow runs.
Subsequent steps
We have launched fundamental snakemake workflows, which provide you with a easy option to run shell instructions in the proper order. snakemake already provides a couple of good enhancements over working the shell instructions by your self or in a shell script –
- it would not run shell instructions if you have already got all of the information you want
- it enables you to keep away from typing the identical filenames time and again
- it offers easy, clear errors when one thing fails
Whereas this performance is good, there are various extra issues we are able to do to enhance the effectivity of our bioinformatics!
Within the subsequent part, we’ll discover
- writing extra generic guidelines utilizing wildcards;
- typing fewer filenames through the use of extra templates;
- offering an inventory of default output information to provide;
- working instructions in parallel on a single laptop
- loading lists of filenames from spreadsheets
- configuring workflows with enter information